


Story of Discovery: The NIDDK provides foundational research support for first FDA-approved therapy to treat the metabolic disease primary hyperoxaluria type 1
Research Update
Primary Hyperoxaluria (PH)
PH is a set of genetic metabolic disorders characterized by increased levels of oxalate in the kidneys, urine, and other organs of the body. The three types of PH (PH1, PH2, and PH3) are caused by a "protein deficiency" and distinguished by deficiencies in different proteins.
Primary Hyperoxaluria Type 1 (PH1)
- Primary hyperoxaluria type 1 (PH1) is a rare disorder that mainly affects the kidneys. It results from buildup of a substance called oxalate, which is normally filtered through the kidneys and excreted in the urine. In people with PH1, the accumulated oxalate is deposited in the kidneys and urinary tract and combines with calcium to form calcium oxalate— the main component of kidney stones. Symptoms of kidney stones include sudden abdominal or flank pain, blood in the urine, frequent urge to urinate, pain while urinating, and/or fever and chills.
- Indications and symptoms of PH1 vary in severity and may begin any time from infancy to early adulthood. People with PH1 may experience recurrent kidney stones, blood in the urine, and urinary tract infections. PH1 can result in end- stage kidney disease, which is life-threatening, and the need for dialysis. As kidney function worsens, oxalate can build up and damage other organs, including the heart, bones, and eyes. Treatments for the disease have been limited, and the only effective treatment for most people is a combined liver- kidney transplant. The lack of effective, nonsurgical treatment options underscores the urgent need to develop new drug therapies for this serious disease.
Genetic Mechanisms of PH1
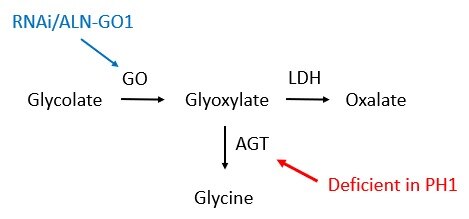
- PH1 is caused by mutations in a gene called AGXT. This gene gives the body instructions for producing a protein called alanine-glyoxylate aminotransferase (AGT). AGT is found in the liver and converts a compound called glyoxylate to the amino acid glycine.
- Mutations in the AGXT gene lead to a deficiency of AGT to convert glyoxylate to glycine. This, in turn, causes glyoxylate to accumulate, and it is ultimately converted to oxalate by a protein called lactate dehydrogenase. Excess oxalate that is not excreted from the body then combines with calcium to form calcium oxalate, leading to kidney damage.
- The metabolic pathway leading to oxalate production includes the conversion of glycolate to glyoxylate by the protein glycolate oxidase (GO).
- Thus, glycolate oxidase is an important player in the biological mechanism leading to high levels of oxalate in PH1. Researchers discovered that targeting and thus reducing levels of glycolate oxidase using RNA interference (RNAi) technology was a promising strategy to reduce oxalate levels and treat the disease.
The Research Path to an FDA-approved Treatment
- The accompanying timeline shows that, for many decades, the NIDDK and the NIH have supported foundational research to better understand the metabolic dysfunction underlying PH1 (1960s), including biological pathways involved in oxalate production (1970s), development of RNAi technology to target/silence genes (1990s, 2000s), identifying glycolate oxidase as a promising target to treat PH1 (2000s), and translational research using RNAi-based approaches to target GO and reduce oxalate production in animal models of PH1 (2010s). This research laid the foundation for industry-supported trials of the RNAi therapeutic ALN-GO1 (2010s), culminating in its recent U.S. Food and Drug Administration (FDA) approval as the first drug to treat PH1 in both children and adults (2020). This decades-long story elegantly showcases how basic research elucidated biological mechanisms of both health and disease and resulted in a new therapeutic that greatly improves the prognosis of people with PH1.
- The NIDDK continues its support of this research area to improve the lives of people living with PH1 as evidenced by a recent report describing three patients with PH1 and kidney failure who were able to regain kidney function and discontinue dialysis after treatment with pyridoxine (vitamin B6).
Share this page