About Our Research
The broad goal of our work is the development of novel simulation methodology and theory for better understanding protein function and evolution. Specifically, we are interested in following interrelated topics: the molecular mechanisms of protein folding, protein misfolding and protein aggregation, and in the properties and function of intrinsically disordered proteins. We are also interested in the evolution of sequences which fold to stable structures, and in the design of new sequences with propensities for multiple structures. Our aim is to help to interpret experiments and to answer questions which would be very difficult to directly address experimentally. One of the challenges in studying these problems by molecular simulation is the quality of the simulation models themselves, and so we are putting significant effort into improving these models, at various levels of resolution -- in particular by using experimental data as input. New areas include the development of atomistic simulation models, the study of protein folding dynamics, methods to describe membrane protein folding, the assembly of peptides into amyloid fibrils, and formation of protein 'granules' by phase separation.
Recent Work
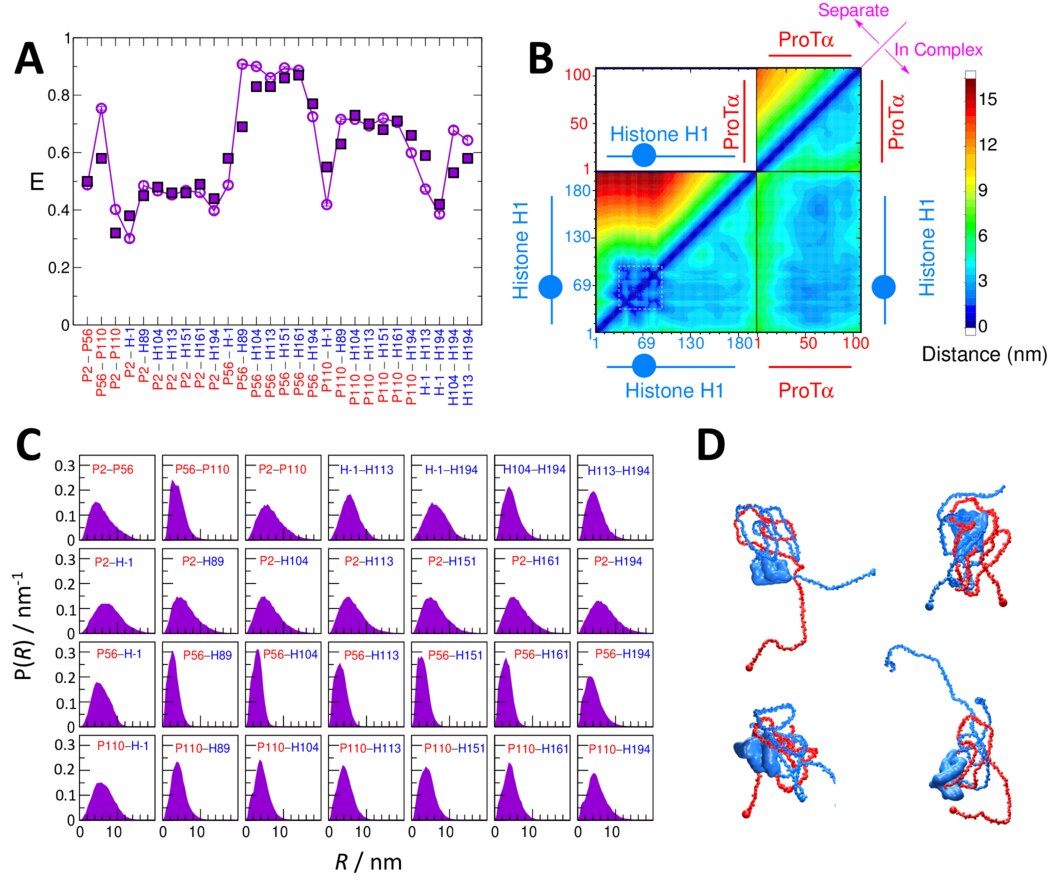
Complexes and assemblies of intrinsically disordered proteins. Association of proteins which do not fold to a specific structure has been found to play important roles in signalling, molecular chaperoning and formation of membraneless compartments within cells. We have developed both atomistic and coarse-grained models to study these processes, and relate the functional mechanism to the properties of the protein sequence.
Cotranslational protein folding. Proteins fold as they are synthesized on ribosomes, but this complexity has only recently been incorporated into experimental and theoretical studies of protein folding. We have collaborated with experimental groups studying protein folding on the ribosome using arrest peptide methodology, in order to provide an interpretation of experimental observations in terms of folding mechanism on the ribosome.
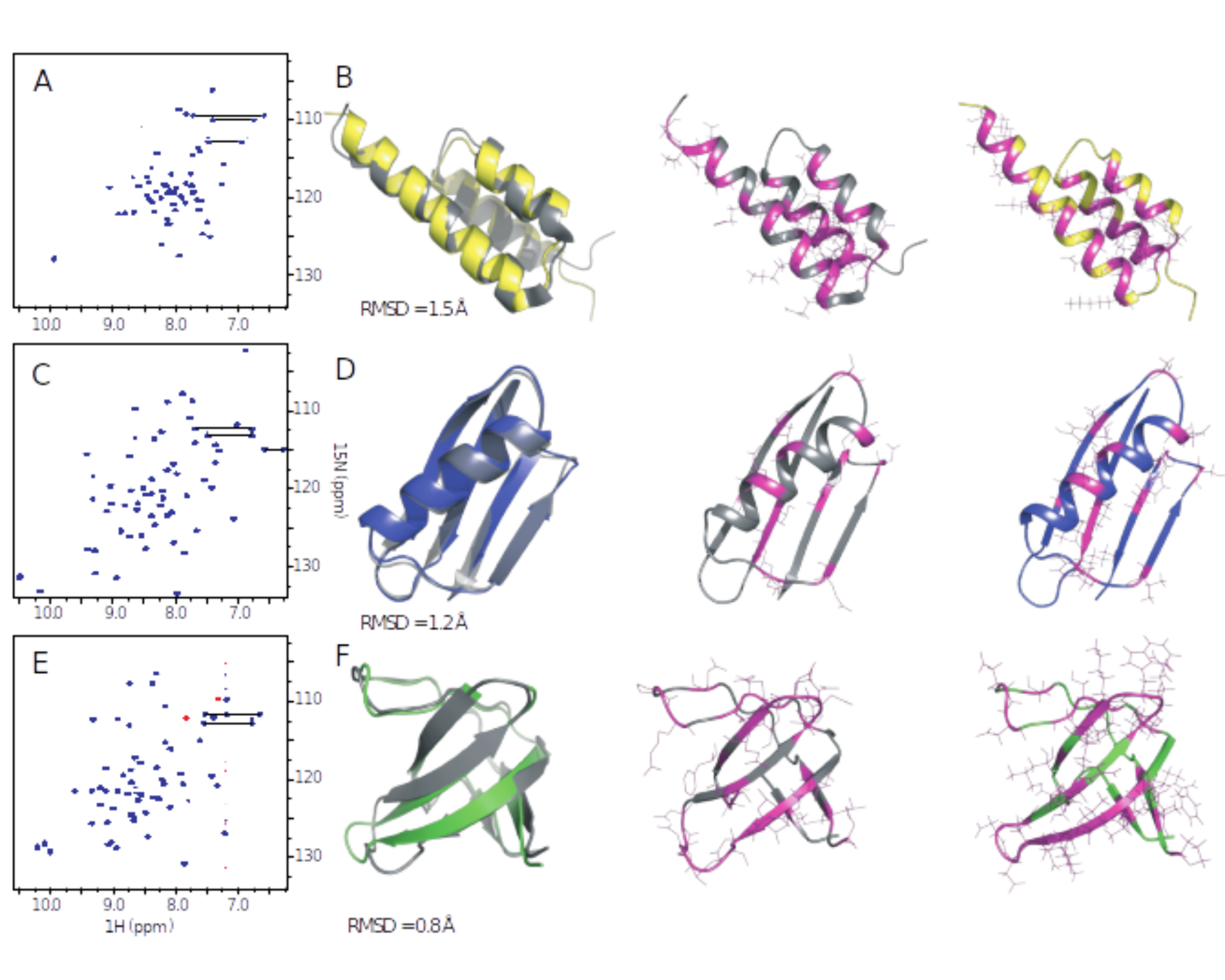
Coevolutionary models for protein folding and fold switching. Coevolutionary models based on sequence data have recently emerged as powerful tools for structure prediction. We demonstrated for the first time that they could also be used to design novel sequences folding to a target structure, for three different protein folds. We are now using a related approach to design novel proteins which can switch between different structures, as well as detect naturally occurring examples of such proteins.
Folding and assembly of membrane proteins. We have studied the assembly of a protypical example of membrane protein assembly, namely the self-association of glycophorin A using both atomistic and coarse-grained models. In doing so we discovered residual deficiencies in the force fields used and devised simple corrections to stabilize the native state. Using transition-path sampling, we were able to describe the assembly mechanism and rates for association and dissociation.
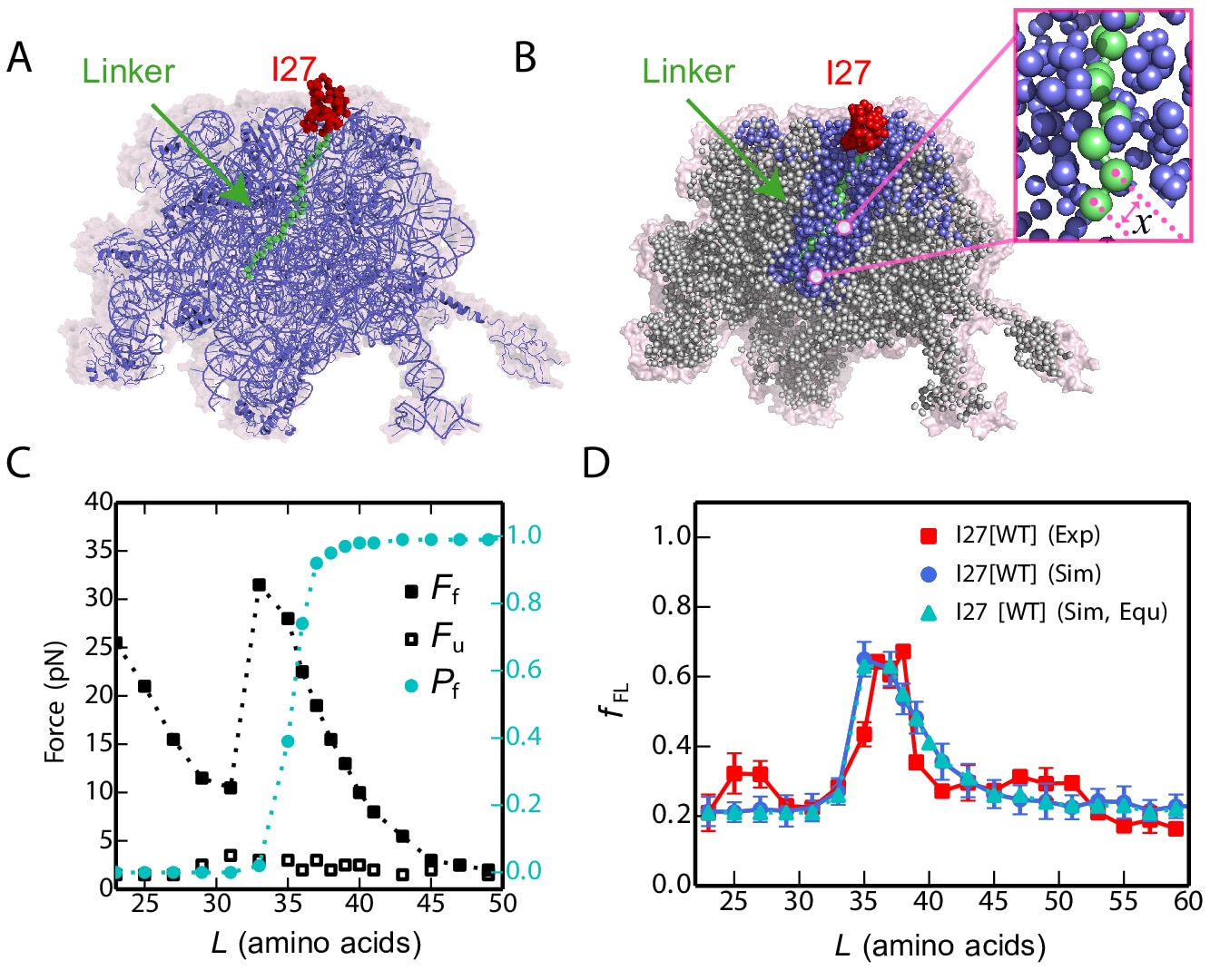
Misfolding in multidomain proteins. Adjacent domains in multidomain proteins frequently have similar structure, raising the question of how they avoid misfolding. We have developed coarse-grained simulation models to help interpret experiments on multidomain misfolding in titin. We have also tried to generalize this model to predict misfolding in other multidomain proteins.
Collapse of unfolded and intrinsically disordered proteins. We have resolved an earlier controversy surrounding discrepancies between the degree of collapse of unfolded and disordered proteins. We were able to show using a close collaboration with experimental groups that these discrepancies arose because of shortcomings in the models used to interpret the data. We have used these insights to develop new models for interpreting FRET and SAXS experiments.
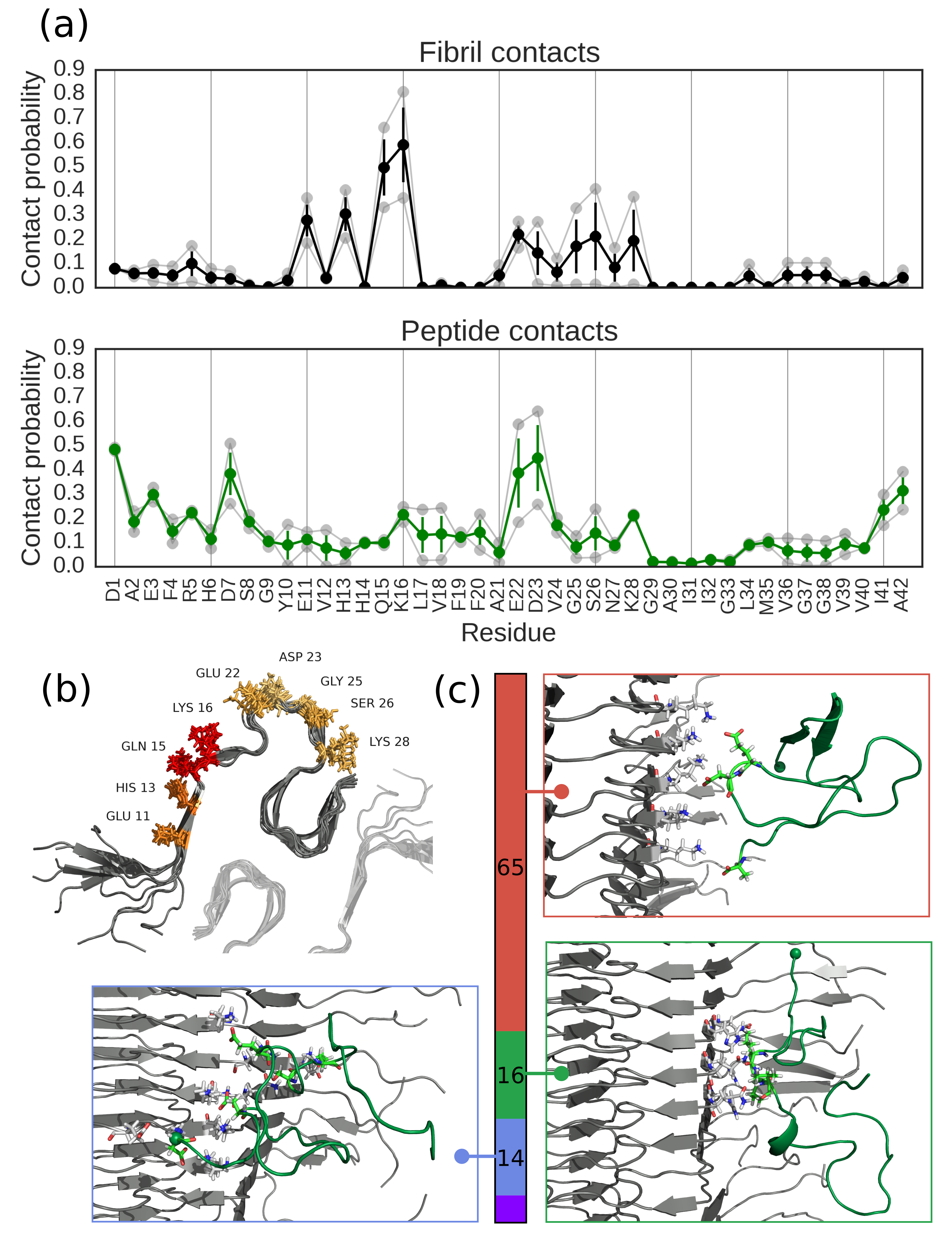
Properties of Alzheimer’s A-beta peptide. We have used all-atom simulations, in conjunction with single molecule FRET to show that the monomer of the Alzheimer’s A-beta peptide is highly disordered, in contrast to earlier results from molecular simulation. We have also used all-atom simulations together with enhanced sampling methods to investigate the association of A-beta42 monomers with the mature fibril structure, providing insights into interactions important for secondary nucleation.
Research Images