About Our Research
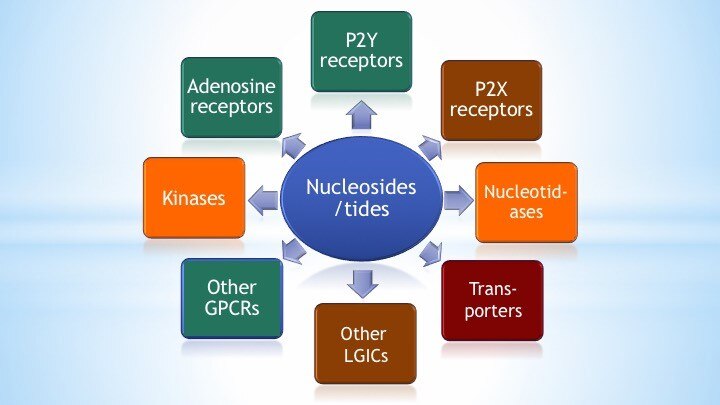
We emphasize four aspects of studying receptors and other proteins in the purinergic signaling pathways: 1) design and synthesis of novel and selective agonists and antagonists based on SAR; 2) protein structure-function studies; 3) exploring the novel biological role of such receptors; and 4) conceptualization of future therapeutics. In this effort, there is a tight coupling of organic synthetic methodology, structural biology, and pharmacology.
Development of Drugs Acting at Adenosine Receptors
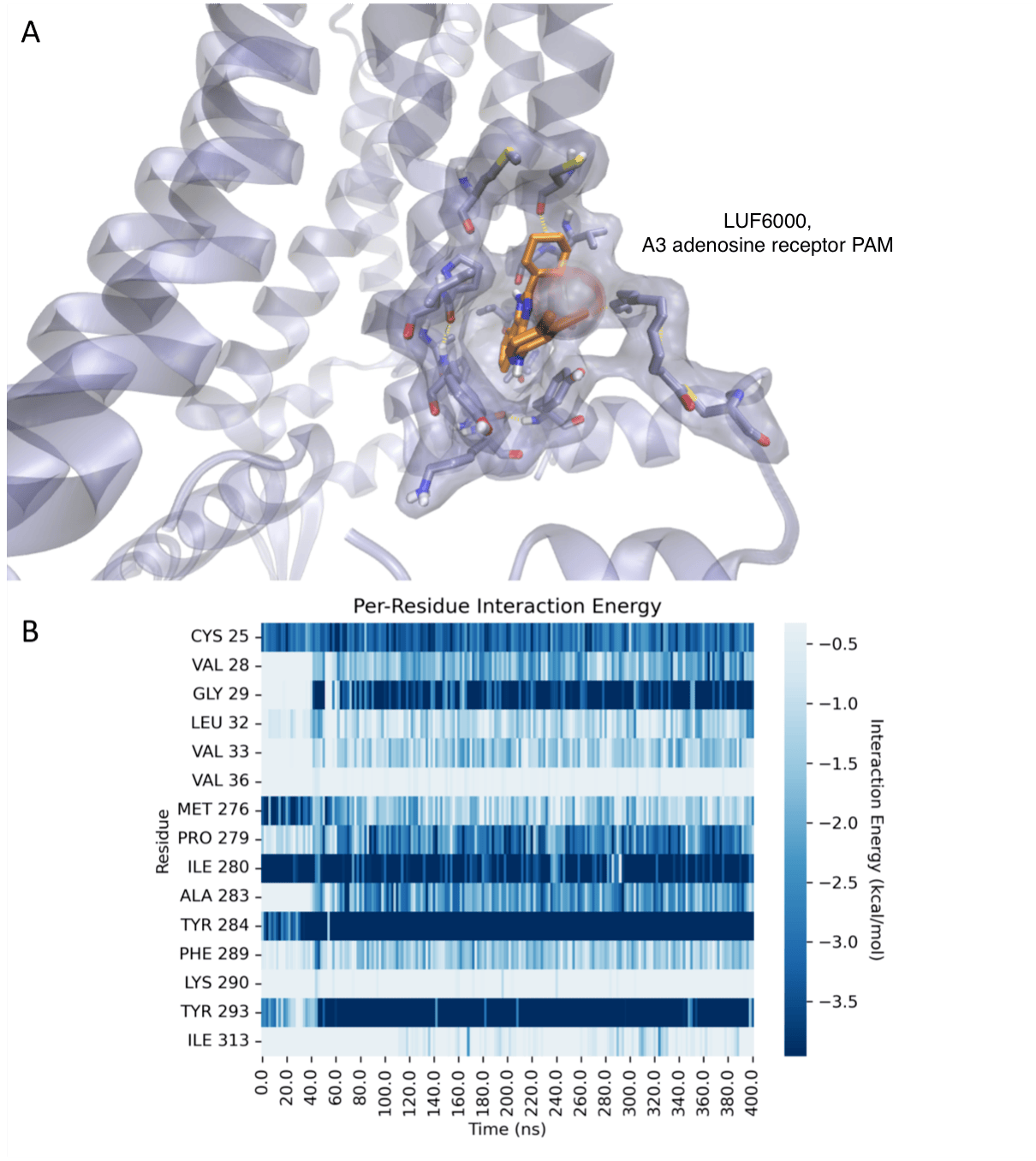
Our overall goal is to design, chemically synthesize, and characterize pharmacologically new agonists and antagonists for the four subtypes of adenosine receptors (ARs), designated A1, A2A, A2B, and A3. Adenosine acts as an important modulator of the activity of every organ in the body and is relevant to a wide range of physiological processes, from the central nervous system, to the immune system and the endocrine system. Adenosine is produced as a metabolite of ATP and is present in the medium surrounding all cells in the basal (resting) state, but its concentration outside the cell rises dramatically when stress to an organ or tissue occurs, such as ischemia, inflammation, and oxidative stress. The ARs are important control elements in responding to physiological stress, often acting as a means of correcting an imbalance in the body, such as delivering more blood to cardiac muscle in distress. The multifold action of adenosine is an endogenous self-correcting system. Thus, adenosine acts as a ubiquitous protective chemical signal in many different scenarios, and selective ligands are being explored as novel therapeutics. For example, agonists for the A3AR appear to be effective in treating autoimmune inflammatory diseases, cancer and neuropathic pain. In our lab, structural leads for novel adenosine receptor modulators are often obtained from known ligands, which can be optimized for selectivity and potency. Structure-based drug discovery is used in this effort. The computer-based screening of chemically diverse libraries to discover new chemotypes is also being used. We aim to understand how changes in the molecular structure of these small molecules alter the receptor subtype selectivity. Screening methodology, such as the introduction of new fluorescent probes and phenotypic in vivo screening, is also being developed.
While compounds that our lab introduced previously are in advanced clinical trials for autoimmune inflammatory disorders (rheumatoid arthritis, psoriasis) and primary liver cancer, the current focus in our lab is the design of novel A3AR agonists for treatment of chronic neuropathic pain, neurodegeneration and other CNS disorders. Our study of chronic neuropathic pain is in collaboration with Daniela Salvemini (St. Louis Univ.). Neuropathic pain conditions are often severe and very difficult to treat, with existing drugs being at least partially effective in only about 50% of the cases. Thus, this represents an important unmet medical need that affects an estimated 100 million people in the US alone.
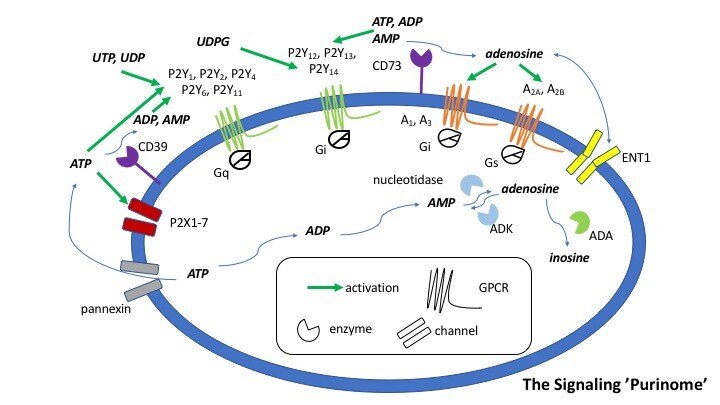
Development of P2Y Receptor Ligands
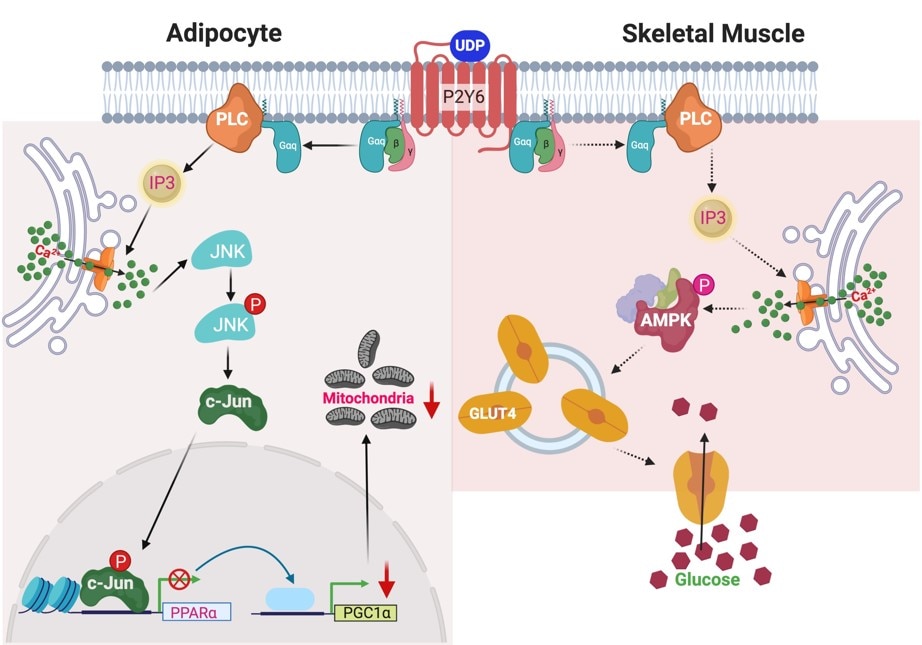
Our overall goal is to design, chemically synthesize, and characterize pharmacologically new ligands for P2Y nucleotide receptors. Both agonists (nucleotide derivatives) and antagonists (nucleotide and nonnucleotide derivatives) are included in this project. Leads will be obtained from known ligands, which can be optimized for selectivity and potency, and from chemically diverse libraries. We will understand how changes in molecular structure of these small molecules alter the receptor subtype selectivity. Structure-based drug discovery, initially based on receptor homology modeling and later on anticipated X-ray crystallographic structure determination for one of the P2YRs structure (collaboration with Ray Stevens, Scripps Res. Inst. and colleagues), are used in this effort. We currently focus primarily on P2Y1, P2Y6, P2Y12 and P2Y14R subtypes. Selective ligands are being used as pharmacological probes to determine the role of extracellular nucleotides in the brain and in peripheral tissues. Our selective ligands for various P2YRs have recently made possible the identification of new therapeutic directions, such as P2Y1 antagonists as antithrombotics, P2Y2 agonists for cardioprotection and antagonists for cancer and inflammation, P2Y6 ligands for neurodegenerative diseases and diabetes, and P2Y14 ligands for inflammation and asthma. Collaborations include studies of the role of P2Y receptors in platelet aggregation, microglial cells, pancreatic islet cells, pain signaling, the immune system, kidney, lung and in muscle cells. The cytoprotective role of UTP, UDP, and other nucleotide analogues in the heart, skeletal muscle, pancreas, and other tissues is to be further explored. The development of effective radioligands and fluorescent probes for the P2Y receptors is also a goal of this project. Type 2 Diabetes (T2D) is becoming a global epidemic and is afflicting the growing proportion of the population. We are studying the purinergic mechanisms regulating function of key metabolic tissues such as adipose tissue and skeletal muscle.
Development of Drugs Acting at Ion Channels
We seek to understand how changes in the molecular structures of activators and antagonists of ligand-gated ion channels and transporters affect their biological activity. We aim to design versatile organic synthetic approaches to the preparation of analogues of such agents. The new analogues will be examined for biological action in cell systems representative of action in the cardiovascular, nervous, pulmonary, and inflammatory systems. Selected compounds are to be studied in vivo by collaborators. New approaches toward pain-control and neuroprotection by modulation of channel are being explored. Activators of ATP-gated ion channels (P2X receptors, P2XRs) are being synthesized and investigated for cardioprotection. We are designing and synthesizing P2X4 agonists (nucleotide derivatives) as cardioprotective agents. We are also interested in antagonists of P2X4R and other P2XR subtypes, as cerebroprotective agents. The dopamine transporter is relevant to behavioral disorders and addiction. We have discovered rigid nucleosides that are allosteric modulators of the dopamine transporter (a sodium symporter) and we are exploring their structure activity relationship. They both enhance binding of cocaine-like molecules and inhibit the transport of dopamine.
Structural Probing of G Protein-Coupled Receptors
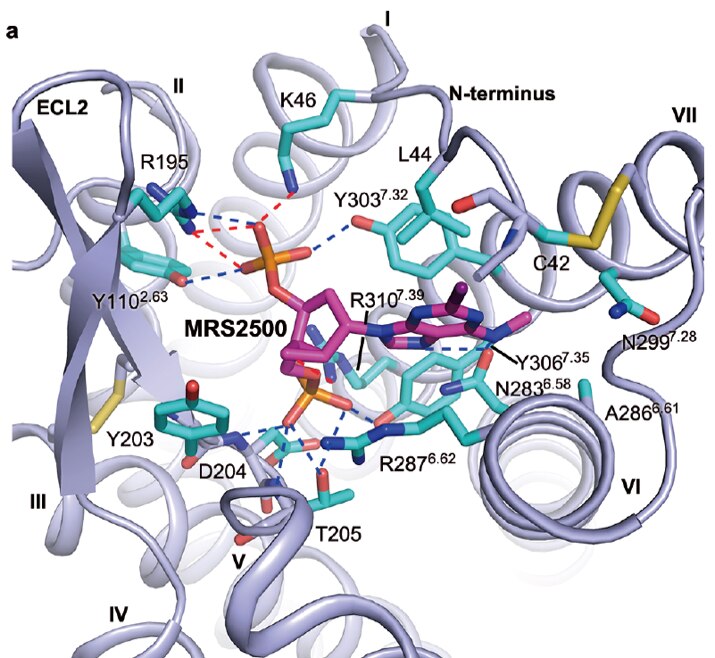
Recent crystallographic structures of G protein-coupled receptors (GPCRs) have greatly advanced our understanding of the recognition of their diverse agonist and antagonist ligands. In additional to structure determination through collaborations, an objective of this project is to explore three-dimensional structures of other GPCRs not yet determined, based on homology to known structures (X-ray and cryo-electron microscopy), such as the A2A adenosine receptor (A2AAR, in both agonist- and antagonist-bound states). Models of the ligand binding site and conformational changes involved in activation of the receptors are being explored using diverse sources of structural data, including mutagenesis and structure activity relationships of ligands. We have applied this approach to the A1 and A3ARs and to the P2Y6 and P2Y14 receptors (P2YRs) for nucleotides. The efficiency of the virtual screening and rational lead optimization tools in harnessing active-state GPCR structures for discovery of novel agonists needs to be explored. Thus, using ligand docking and molecular dynamics simulations for a given GPCR, we aim to investigate the bound conformations and the basis for affinity, selectivity and (where present) the functional activity of a ligand (including biased agonism). Virtual chemical libraries are screened in silico for the discovery of novel ligands.
Functionalized Congeners of Bioactive Compounds
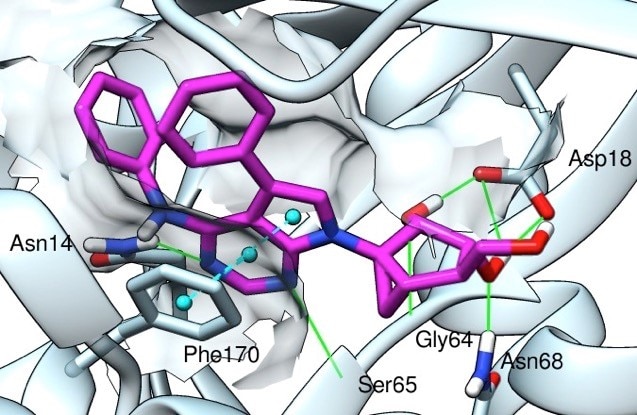
We are using chemical synthesis and pharmacology to explore the structure activity relationships of ligands of G protein-coupled receptors (GPCRs) and other proteins in the purinergic signaling pathways. Nonpeptide small molecule agonists generally bind within the heptahelical transmembrane domain of a receptor to trigger intracellular signaling pathways. An approach to designing ligands for GPCRs by applying structural modification at their permissive distal sites has led to the concept of functionalized congeners. The development of functionalized congeners for a given receptor involves exploring the surface contact of distal structural units of the bound ligand, away from the pharmacophore region, with a GPCR at/near its extracellular domain. These secondary interactions may impart additional selectivity and affinity. This approach enables ligands for GPCRs to be attached to carrier molecules for delivery or reporter groups for detection, and the chemical functional groups and geometry of a linker moiety can be varied as guided by receptor affinity. Multivalent ligands for the activation of GPCRs are being explored. Fluorescent conjugates of agonists and antagonists of high affinity that were synthesized in our lab have been shown to be useful in characterizing receptor binding and for drug screening. For example, we have designed and synthesized functionalized congeners of inhibitors (or antagonists) of the A2A adenosine receptor and the enzyme CD73, which are targets for cancer and neurodegeneration). Functionalized congeners of adenosine receptor ligands (i.e. both agonists and antagonists) are being derivatized for coupling to nanoparticles, such as quantum dots and gold particles, for tissue-specific targeting.
Research Images